Darolutamide observational study in patients with non-metastatic castration-resistant prostate cancer
Geoffrey Gotto1, Evan Y. Yu2, Christopher Pieczonka3, Andrew J. Armstrong4, Hiroyoshi Suzuki5, James Bailen6, Declan Murphy7, Thierry Lebret8, Murilo Luz9, Antoine Thiery-Vuillemin10, Jorge A. Ortiz11, Javeed Khan12, Alberto Briganti13.
1Southern Alberta Institute of Urology, University of Calgary, Calgary, AB, Canada; 2University of Washington, Seattle, WA, United States; 3Associated Medical Professionals of NY, Syracuse, NY, United States; 4Duke University School of Medicine, Durham, NC, United States; 5Toho University Sakura Medical Center, Chiba, Japan; 6First Urology PSC, Jeffersonville, IN, United States; 7Peter MacCallum Cancer Centre, University of Melbourne, Melbourne, Australia; 8Hôpital Foch, Université Versailles St Quentin, Paris, France; 9Hospital Erasto Gaertner, Curitiba, Brazil; 10Centre Hospitalier Régional Universitaire, Besançon, France; 11Bayer Healthcare, Whippany, NJ, United States; 12Bayer Healthcare, Reading, United Kingdom; 13Urological Research Institute, IRCCS Ospedale San Raffaele and Università Vita-Salute San Raffaele, Milan, Italy
Introduction: In ARAMIS (NCT2200614), darolutamide (DARO) improved metastasis-free survival and overall survival in non-metastatic castration-resistant prostate cancer (nmCRPC), with favorable tolerability. The DARolutamide ObservationaL (DAROL) trial (NCT04122976) is assessing real-world safety and effectiveness of DARO. We report the first interim analysis.
Methods: DAROL is an international, single-arm, non-interventional study. Eligible patients (pts) are DARO-naive, ≥18 years of age, with confirmed nmCRPC. Treatment dose/duration are per investigator’s routine practice. The primary endpoint is safety. Prostate-specific antigen (PSA) response is an exploratory objective. Descriptive statistics are reported for the first prespecified interim analysis after ~100 pts (Canada, Japan, U.S.) completed ≥6 months of treatment/discontinued treatment.
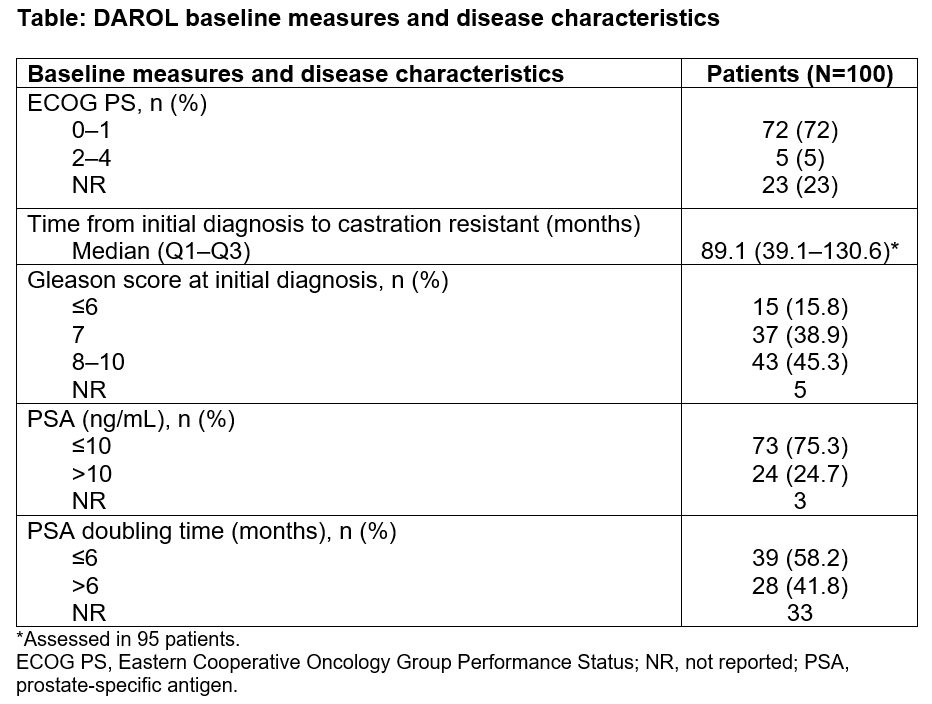
Results: All 100 pts were evaluable for safety: median age 78.0 years; White/Black/Asian/other/not reported 59%/11%/27%/1%/2%; Canada/Japan/U.S. 24%/21%/55% (see baseline measures in Table 1). Most (79%) pts had ≥1 concomitant medication; those in ≥15% were HMG-CoA reductase inhibitors (30%), bone structure/mineralization agents (22%), oral platelet aggregation inhibitors (21%), angiotensin II receptor blockers (16%), alpha-adrenoreceptor antagonists (15%), and beta-blocking agents-antimigraine (15%). Median (Q1–Q3) DARO treatment duration was 11.3 months (8.4–14.4); median (Q1–Q3) followup time was 12.3 months (9.6–15.6). Twenty-eight percent had any grade treatment-emergent adverse events (TEAEs): 3% grade 3, no grade ≥4; 2% had serious adverse events (AEs). Incidence of AEs was low; those in ≥2% of pts were fatigue (5%), diarrhea (5%), asthenia (2%), muscle weakness (2%), anemia (2%), and rash (2%). TEAEs led to drug discontinuation in 7%. Of 93 pts, 81.7%, 77.4%, and 52.7% had PSA responses of ≥30%, ≥50%, and ≥90% (any time during followup), respectively.
Conclusions: Safety and tolerability of DARO were consistent with the favorable tolerability profile observed in ARAMIS.